




近日,中国农业科学院深圳农业基因组研究所(岭南现代农业科学与技术广东省实验室深圳分中心)易会广课题组在《生物信息学(Bioinformatics)》上发表了题为“Kssdtree:一个基于sketching技术的系统发育分析交互式Python包(Kssdtree: an interactive Python package for phylogenetic analysis based on sketching technique )”的论文,该研究开发了一个基于sketching技术的Python包用于系统发育分析,能够准确和快速的构建系统发育树。
新一代测序技术的出现显著增加了基因组数据的体量和多样性,对分析方法提出了更高的效率要求,传统的系统发育分析方法依赖于序列比对工具(如BLAST、CLUSTAL和MUSCLE),非常耗时,耗资源;尽管非比对方法(如co-phylog和AAF)提供了快速处理数十个细菌基因组的能力,但在处理大规模细菌基因组或中等规模的大型真核基因组时仍然面临挑战;最近,基于sketching技术成为实时、大规模系统发育分析的一种有前景的解决方案。然而,现有的基于sketching的系统发育工具(如Mashtree)存在缺陷,包括平台限制、缺乏可视化和固有的距离估计偏差,这些限制共同阻碍了分析的便利和效率。
因此,研究人员开发了一个基于sketching的系统发育分析Python包Kssdtree,有效地解决了平台限制、可视化以及距离估计偏差问题。Kssdtree为特定类型的系统发育分析提供了三种不同的流程(图1) :常规流程、参考减法流程和基于GTDB的系统发育定位流程。常规流程提供了通用系统基因组发育分析。参考减法流程设计用于种内基因组聚类分析。基于GTDB的系统发育定位流程允许从GTDB数据库中检索最相似的基因组,并使用检索到的基因组与查询基因组一起执行系统发育分析。
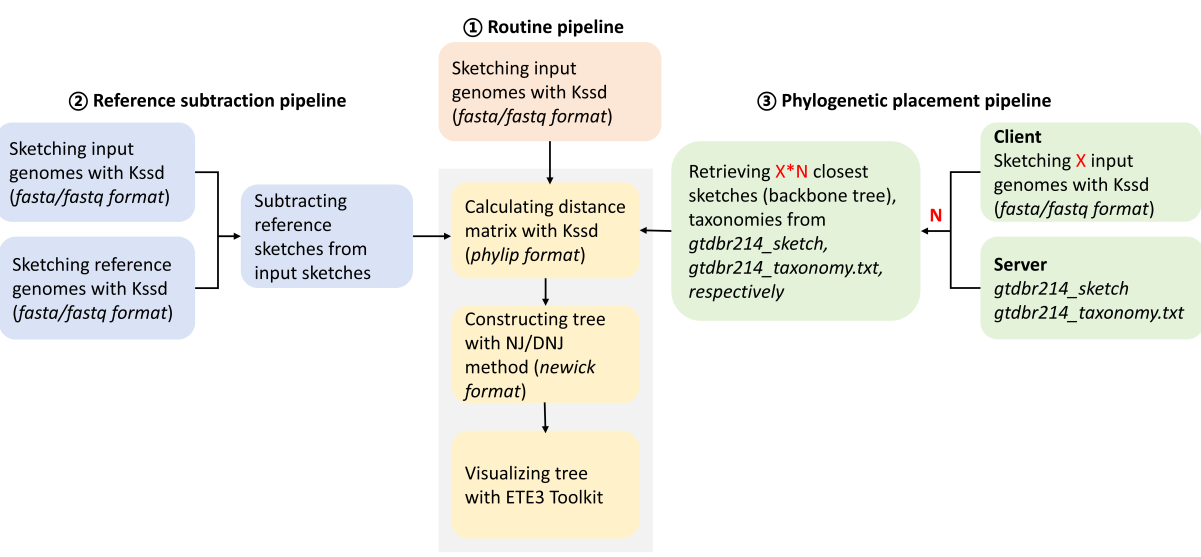
图1 | Kssdtree工作流程
研究对比了Kssdtree和其他基于sketching的方法(Mashtree、BinDash+NJ/DNJ、 SourMash+NJ/DNJ)的准确性和时间效率。研究收集了真实和模拟的基准测试数据集。真实数据集包括29个大肠杆菌/志贺氏菌基因组(ES29)、25个鱼类线粒体基因组(FM25)和14个植物物种(P14)。模拟数据集由ALF模拟工具产生的200个物种组成。此外,使用DWGSIM测序数据模拟工具生成四个数据集的未组装的原始测序数据。为了对运行时间效率进行基准测试,从最新的NCBI细菌RefSeq数据库中随机抽取1000、5000和10000个细菌基因组,分别表示为BACT1000、BACT5000和BACT10000。Kssdtree在综合基准测试数据集的准确性和时间效率方面表现出卓越的性能,超越了其他基于sketching的工具。
研究对种内系统基因组分析进行基准分析,从最近发表的人类泛基因组参考文献中下载了43个相对完整的人类基因组。结果表明,其他基于sketching的方法倾向于基于性别对基因组进行聚类。这是因为人类基因组之间的差异主要来自性染色体,而这些方法不支持参考减法操作。相反,Kssdtree能够进行草图减法操作,在距离计算和树构建之前,从单个基因组草图中减去参考草图(包括性染色体)。这种方法允许Kssdtree根据种群明确地组织基因组,而不考虑性别影响(图2)。
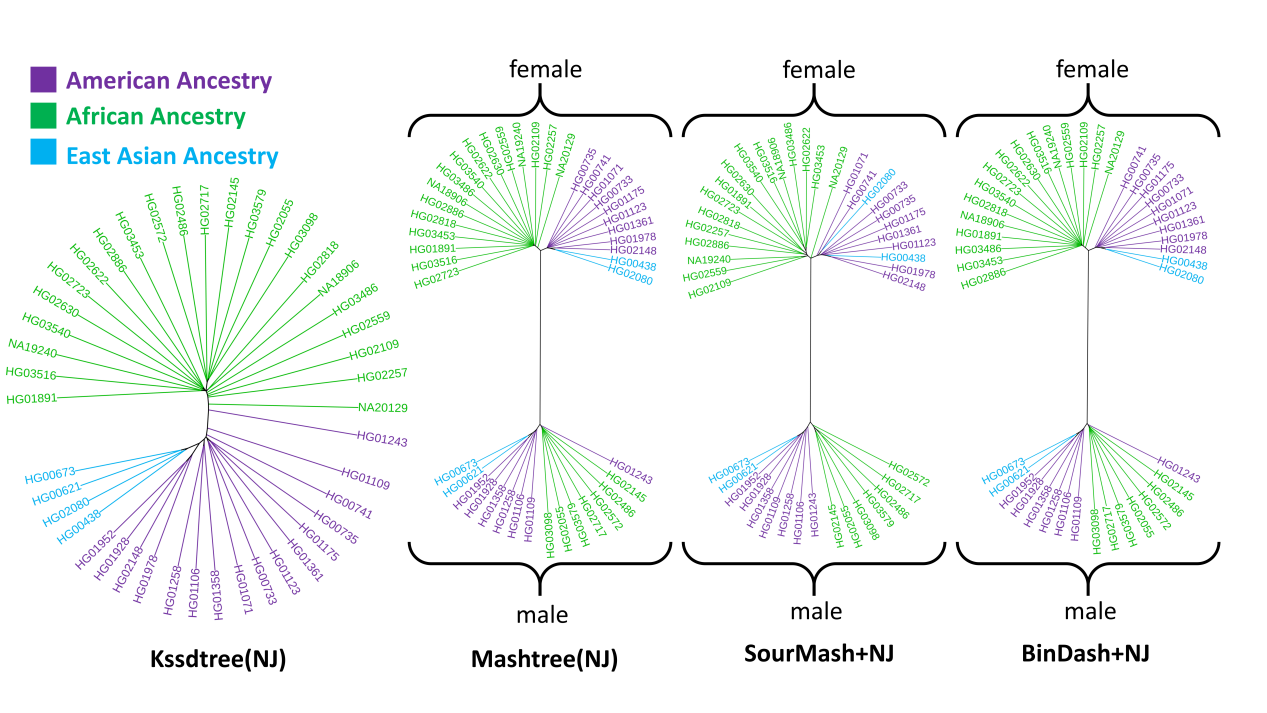
图2 | 比较不同方法构建的43个人类基因组系统发育树
研究还使用大肠弯曲杆菌的组装基因组和未组装的原始reads来测试基于GTDB的Kssdtree系统发育定位。Kssdtree基于检索到的最相似基因组的分类,正确地识别了查询基因组的起源物种。查询基因组与GTDB中最相似的基因组之间的系统发育关系(图3),使我们能够使用GTDB数据库确定查询基因组在原核生命树中的系统发育位置。
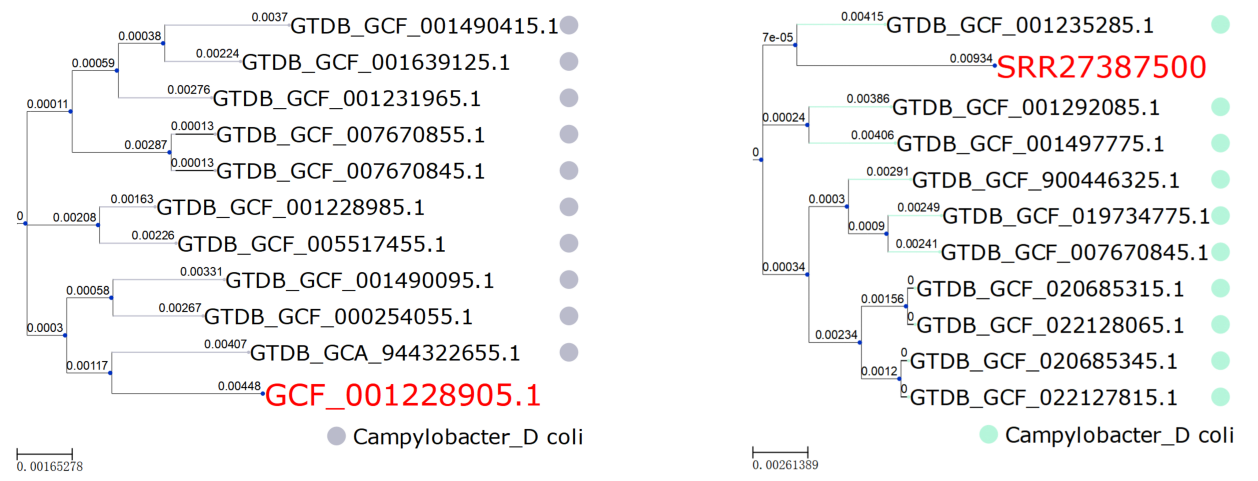
图3 | Kssdtree对Campylobacter coli物种的组装基因组GCF_001228905.1和原始测序Run SRR27387500进行系统发育定位分析
值得注意的是,Kssdtree提供了诸如多平台支持(Windows、Linux和MacOS)、树可视化、种内系统发育分析和基于GTDB的系统发育定位分析等关键优势,显著提高了系统发育研究的范围和深度。
基因组所与太原理工大学联培硕士杨航为论文第一作者,基因组所易会广副研究员、太原理工大学郑文教授和陈泽华教授为论文的共同通讯作者。基因组所与太原理工大学联培硕士畅佳星,基因组所易会广课题组科研助理卢晓欣、常青(已离职)参与了该研究。该项目得到深圳市基础研究机构、深圳市基础研究计划、深圳市博士后出站科研资助和深圳市大鹏新区博士后出站科研资助。
原文链接:https://doi.org/10.1093/bioinformatics/btae566
